Published Date
Abstract










For further details log on website :
http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0122123
- Published: August 30, 2013
- http://dx.doi.org/10.1371/journal.pone.0074736
Author
Abstract
Coconut, a member of the palm family (Arecaceae), is one of the most economically important trees used by mankind. Despite its diverse morphology, coconut is recognized taxonomically as only a single species (Cocos nucifera L.). There are two major coconut varieties, tall and dwarf, the latter of which displays traits resulting from selection by humans. We report here the complete chloroplast (cp) genome of a dwarf coconut plant, and describe the gene content and organization, inverted repeat fluctuations, repeated sequence structure, and occurrence of RNA editing. Phylogenetic relationships of monocots were inferred based on 47 chloroplast protein-coding genes. Potential nodes for events of gene duplication and pseudogenization related to inverted repeat fluctuation were mapped onto the tree using parsimony criteria. We compare our findings with those from other palm species for which complete cp genome sequences are available.
Figures
Citation: Huang Y-Y, Matzke AJM, Matzke M (2013) Complete Sequence and Comparative Analysis of the Chloroplast Genome of Coconut Palm (Cocos nucifera). PLoS ONE 8(8): e74736. doi:10.1371/journal.pone.0074736
Editor: Hector Candela, Universidad Miguel Hernández de Elche, Spain
Received: June 25, 2013; Accepted: August 6, 2013; Published: August 30, 2013
Copyright: © 2013 Huang et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was funded by Academia Sinica (http://www.sinica.edu.tw/main_e.shtml). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have declared that no competing interests exist.
Introduction
Chloroplasts (cp) are cell organelles that carry out photosynthesis, thus converting light energy into chemical energy in green plants and algae. Chloroplasts contain their own genome, which in flowering plants usually consists of a circular double-stranded DNA molecule ranging from 120 to 160 kb in length [1]. The cp genome is divided into four parts comprising a large single copy region (LSC) and a small single copy region (SSC), which are separated by a pair of inverted repeats (IRs). Cp genomes typically encode four rRNAs, around 30 tRNAs and up to 80 unique proteins [2]–[4].
With the advent of high-throughput sequencing technologies and their use in obtaining complete plastid genomes [5], [6], the number of fully sequenced cp genomes has increased rapidly. To date, the Complete Organelle Genome Sequences Database (http://amoebidia.bcm.umontreal.ca/pg-gobase/complete_genome/ogmp.html) lists 324 complete cp genome sequences spanning 268 distinct organisms. The complete cp genome sequences include date palm (Phoenix dactylifera L.) and oil palm (Elaeis guineensis Jacq.). Both are members of the palm family (Arecaceae), which is the third most economically important family of plants after the grasses and legumes [7]. Complete sequence information on cp genomes from three additional palms - Calamus caryotoides, Pseudophoenix vinifera, Bismarkia nobilis – has recently been deposited in GenBank [8]. However, the complete cp genome sequence of coconut palm (Cocos nucifera L.), which is a universal symbol of the tropics and equally important as oil palm [7], has not yet been reported.
Coconut is one of the most important crops in tropical zones where it is a source of food, drink, fuel, medicines and construction material [9]. In addition, coconut oil is used for cooking and for pharmaceutical and industrial applications [10]. Although coconut trees display considerable morphological diversity, they are considered taxonomically a single species (and the only species) within the genus Cocos. Based on stature and breeding, coconut cultivars can be divided into two groups: tall and dwarf [11]. The former typically grows up to 35 to 40 meters and is mainly outcrossing, whereas the latter can only grow up to 25 to 30 meters and usually is selfing. Dwarf coconuts, which are less common than the tall variety, are usually found growing close to humans and have traits that likely result from human selection [10]. Here we report the complete cp genome sequence of a dwarf coconut plant, which is thought to be descended from coconut trees originally imported into Taiwan from Thailand (personal communication from private breeder).
Materials and Methods
Whole genome sequencing and de novo assembly
Fresh young leaf material (ca. 2 g) was collected from a coconut seedling growing under ambient conditions in the greenhouse of Academia Sinica and the genomic DNA (gDNA) was extracted using a modified CTAB protocol [12]. We used the ratio of absorbance at 260 nm and 280 nm (A260/280) and gel electrophoresis to measure the purity and integrity of the extracted gDNA. High quality DNA (concentration >100 ng/µl; A260/230>1.7; A260/280 = 1.8∼2.0) was sequenced using the Illumina GAIIx platform (YOURGENE BIO SCIENCE Co., New Taipei City, Taiwan). Short reads (70 bp) from paired-end sequencing were trimmed with a 0.05 error probability. The trimmed reads were de novo assembled using CLC Genomic Workbench 6.0.1 (CLC Bio, Aarhus, Denmark). The de Bruijn Graph approach with a k-mer length of 22 bp and a coverage cutoff value of 10X was applied for assembly. The average read length and insert size were 151 bp and 340 bp respectively. The assembled contigs shorter than 200 bp were removed from the scaffold while those with coverage larger than 10X were selected for BLAST search against plastid genomes of date palm [2], oil palm [3], and other chloroplast sequences with an e-value cutoff of 10−5 (199 sequences in total). Gaps between contigs were filled by PCR amplification with specific primers that were designed based on contig sequences or homologous sequence alignments (Table S1). The PCR products were purified with GEL/PCR DNA clean-up kit (Favorgen Biotech Corp.) and then sequenced by conventional Sanger sequencing. The sequencing data along with gene annotation have been submitted to GenBank with an Accession number of KF285453.
Genome annotation, base composition, repeat structure, and codon usage
Preliminarily gene annotation was carried out through the online program DOGMA [13] and BLAST searches. To verify the exact gene and exon boundaries, we used MUSCLE [14] to align putative gene sequences with their homologues acquired from BLAST searches in GenBank. All tRNA genes were further confirmed through online tRNAscan-SE search server [15]. The online program tandem repeat finder [16] was used to search the locations of repeat sequences (>10 bp in length) with the following set up: (2, 7, 7) for alignment parameters (match, mismatch, indels); 80 for minimum alignment score to report repeat; and maximum period size of 500. Codon usage was calculated for all exons of protein-coding genes (pseudogenes were not calculated). Base composition was calculated by Artemis [17].
Analysis of RNA editing
Potential RNA editing sites in protein-coding genes of coconut cpDNA were predicted by the online program Predictive RNA Editor for Plants (PREP) suite (http://prep.unl.edu/) [18] with a cutoff value of 0.8. This program contains 35 reference genes for detecting RNA editing sites in plastid genomes. The predicted editing sites were verified by reverse transcription polymerase chain reaction (RT-PCR) experiments. In addition to those genes predicted by the program, we also investigated rpl22, rpl23, rps3, rps7, ycf1, ycf2, and ycf4 genes, within which RNA editing sites were reported in the cp genome of oil palm [3]. The Plant Total RNA Miniprep Purification Kit (GMbiolab Co., Ltd.) was applied to extract total RNA from leaf of the same seedling used for DNA extraction. The first strand cDNA was synthesized with QuantiTect Reverse Transcription Kit (Qiagen) following the manufacturer's protocol. Gene specific primers for cDNA amplification were designed based on homologous sequence alignment. Maximum 1 µl of the reaction mixture was used as template for PCR amplification. The PCR products were purified with GEL/PCR DNA clean-up kit (Favorgen Biotech Corp.). Purified PCR products were sequenced using ABI PRISM® 3700. A complete primer list is provided in Table S1.
Phylogenetic analysis
Forty seven protein coding genes were extracted from 25 taxa, including Amborella, Nuphar, 17 species of monocots, four species of magnoliids, and two species of eudicots. The GenBank accession number of each taxon is provided in Table 1. These taxa were selected because they have complete or nearly complete plastid genomes deposited in GenBank. Nucleotide sequences of each gene were first aligned by MUSCLE [14] through the online server of European Bioinformatics Institute (http://www.ebi.ac.uk/Tools/msa/muscle). The aligned sequences were then concatenated through copy and paste in text editor. The statistical method of Maximum Likelihood (ML) and the computer program Garli version 2.0 were applied for phylogenetic reconstruction, with parameters estimated from the data. The GTR substitution model with evolutionary rates among sites evaluated by a discrete gamma distribution was used for tree search. All positions containing gaps or missing data were eliminated. Branch support was evaluated by 1,000 replications of bootstrap (BS) re-sampling.
Table 1. Accessions and references for taxa used in phylogenetic reconstruction and genome comparison in this study.
Results and Discussion
Sequencing and de novo assembly
Illumina sequencing produced 6,413,504 paired-end reads with an average read length of 151 bp and a total base number of 968,439,104. After quality trim, 6,328,120 reads with an average of 145.3 bp and a total base number of 919,475,836 remain. The subsequent de novoassembly and reference-guided blast search resulted in five major contigs separated by five gaps, which were then filled by Sanger sequencing. In addition to gap closure and confirmation of four junction regions (LSC/IRA, LSC/IRB, SSC/IRA, SSC/IRB), we also validated the accuracy of our whole genome sequencing by randomly selecting genes/spacers for PCR-based sequencing. Priority was given to long genes (e.g., ycf1, ycf2, rpoC1) or long spacers (between pairs of rpoB and psbD, ycf2andndhB, ndhC and trnV-UAC). A few regions where genes were transcribed from clockwise to counterclockwise (vice versa) were also validated.
Organization of chloroplast genome
Analysis of the data obtained from high-throughput sequencing demonstrated that the cp genome of coconut is a typical quadripartite molecule (Fig. 1) within which a pair of inverted repeats (IRs) is separated by a large single copy region (LSC) and a small single copy region (SSC). The genome is 154,731 bp in length (IRs = 53,110 bp; LSC = 84,230 bp; SSC = 17,391 bp) and is predicted to encode 130 genes and four pseudogenes. The former includes 84 protein-coding genes, 38 tRNA genes, and eight rRNA genes while the latter is represented by pseudo ycf1, rps19, and two copies of ycf15. Of those genes, three protein-coding genes (ycf2, ndhB, and rps7), four rRNA genes (rrn16, rrn23, rrn4.5, and rrn5), and eight tRNA genes are present in two copies (Fig. 1).
Figure 1. Coconut chloroplast genome map.
Genes shown on the outside of the large circle are transcribed clockwise, while genes shown on the inside are transcribed counterclockwise. Thick lines of the small circle indicate IRs. Genes with intron are marked with “*”. Pseudo genes are marked with “Ψ”.
Fourteen of the protein-coding genes and eight of the tRNA genes contain introns; and four pairs of genes overlap (4 bp between atpE and atpB; 10 bp between ndhK and ndhC; 53 bp between psbC and psbD; and 57 bp between pseudo ycf1 and ndhF). Each intron-containing gene has only one intron, except ycf3 and clpP, which have two introns. Most protein-coding genes have standard AUG as initiator codon; however, rpl2 and ndhD have an initiator codon of ACG, rps19 starts with a GUG codon, and the initiator codon of cemA is ambiguous. The frequency of codon usage in the coconut cp genome is summarized in Table 2. Similar to many cp genomes of angiosperms [2], [3], [19]–[22], a strong bias toward an A or T in the third position of synonymous codons is also observed in the coconut cp genome. The most and least prevalent amino acids are leucine (2624) and cysteine (323), respectively.
Table 2. Codon usage and codon-anticodon recognition pattern in cp genome of coconut.
Although RT-PCR analysis validated that C-to-U editing changed the ACG start codon to AUG in the ndhD gene, the ACG start codon in the rpl2 gene appeared to remain unedited in repeated experiments. However, we cannot eliminate the possibility that a low level of editing occurs in rpl2. Although less frequent than AUG, translation initiated at an ACG or GTG start codon is not unprecedented in plants. A previous study demonstrated that an initiator codon of AUG is not required to specify the initiation site for a proper translation in the cp genome [23]. GUG codons have been shown to be more efficient than ACG in initiating translation and have a relative strength varying from 15 to 30% of AUG activity [24]. In angiosperms, a GUG start codon has been found in the cemA gene [5], [25]–[27] and rps19 gene [2], [3], [5], [8], [26], [28]–[32]. A transcript starting with an ACG start codon has been observed in the ndhD gene in some species of Nicotiana [33], [34].
Repeats
With a criterion of 100% match in repeat copies, the tandem repeat finder identified 13 sets of repeats that are longer than 10 bp, including eight tandem repeats, three direct repeats, and two inverted repeats (Table 3). Three of the repeats are found in the ycf2 genes, which are in the IR regions. The remaining repeats are found in the LSC region: one at the 3′ end of the rps3gene, seven in spacers, and two in the introns. This repeat content is similar to that found in date palm and oil palm. In fact, five of the repeats found in coconut (No. 2, 3, 6, 11and 12 in Table 3) are shared by both oil palm and date palm, though the copy number may differ. In addition, repeats No. 5 and No. 8 in coconut are shared by oil palm while repeats No. 4 and 13 are shared by date palm.
Table 3. Repeat sequences and their distribution in cpDNA of coconut.
Repetitive sequences in cp genomes may recombine and induce rearrangements [35]–[37], which could play a crucial role in stabilization of cpDNA [38]. Compared with other angiosperms, cp genomes of the palm family generally have fewer and shorter repeats (Table 4). Of the 13 repeats found in coconut cpDNA, the longest is 30 bp. The oil palm cp genome has seven repeats and the longest is 40 bp [3] while date palm has 11 repeats and the longest is 39 bp [2]. By contrast, more than 20 repeats, with the longest extending up to 132 bp, were reported in Poaceae [39], [40]. About 232 repeats, ranging from 30 to 61 bp in length, were reported in Cymbidium orchid [29]. In Citrus, 29 repeats with a range of 30 to 59 bp in length were detected [41]. In the Solanaceae family, as many as 42 repeats, with the most extensive being 56 bp, have been reported [42].The cp genome of Gossypiumhas 54 repeats, with a longest one of 64 bp [43]. In the Geraniaceae family, some cp genomes contain up to 9% (or higher) repetitive DNA [4], [44] and many of the repeats are longer than 100 bp [4].
Table 4. Comparison of repeat numbers and repeat lengths among 16 angiosperms.
In view of the correlation between repetitive DNA content and sequence rearrangement, significant structural rearrangements are likely to be observed in cp genomes rich in repetitive sequences. This idea has been validated in many cases listed above such as Poaceae [35], [39], [40], [42] and Geraniaceae [4], [44]–[46]. Conversely, the relatively low content of repetitive DNA in cp genomes of the palm family suggests a relatively higher degree of stability and conservation across different palm species. Consistent with this notion, our investigation revealed neither significant recombination (Fig. S1) nor dramatic variation (Table 5) in the cp genomes of six palm species.
Table 5. Comparison of cp genomes among six palm species.
RNA editing sites
RNA editing is a posttranscriptional process that is mainly observed in mitochondrial and cp genomes of higher plants [47]. This process may induce the occurrence of substitution or indels, which in turn, can result in transcript alternation [33], [47], [48]. In coconut cpDNA, the PREP-cp program predicted 83 RNA editing sites out of 27 genes. Our RT-PCR analysis confirmed editing at 64 of those sites (Table 6). An additional six editing sites not predicted by the program were detected in accD, matK, ndhB, ndhG, ndhH, and rpoA. Of the genes investigated, ndh genes have the highest number of editing sites.
Table 6. RNA editing predicted by PREP-cp program and confirmed by RT-PCR.
The editing types in coconut were all non-silent and 100% C-to-U. One occurrence of editing altered the initiator codon ACG to AUG in ndhD gene. Of these editing events, 62 (82.67%) occurred at the second base of the codon, 12 (16%) were at the first base of the codon, and only one (1.33%) was at the third base of the codon. The conversions of amino acids include 63 hydrophilic to hydrophobic (S to L, S to F, H to Y, T to M, R to W, T to I, and D to F), 11 hydrophobic to hydrophobic (P to L), and one hydrophobic to hydrophilic (P to S).
A comparative study of RNA editing across eight land plants demonstrated an evolutionary trend of decline (or complete loss) in the number of editing sites, silent editing, editing in the first or third position, and editing types other than C to U [47].
In angiosperms, the editing is almost exclusively a C to U substitution [49] and the total number of editing sites ranges from 20 to 37 [47], [50]–[53]. Compared with other angiosperms, coconut has more than twice as many editing sites, although the editing characteristics are similar (Table 7). Moreover, because of the evolutionary conservation of RNA editing, closely related taxa usually share more editing sites [47]. For example, more editing sites are shared within Poaceae than those shared among grasses and dicots [54]. Similarly, related Nicotiana species share more editing sites with each other than with plants from other genera [34].
Table 7. Comparison of RNA editing in six species of angiosperms.
The rps19 pseudogenization and IR fluctuation
Dot plot analysis demonstrated that the gene content and organization of coconut cpDNA are nearly identical to other palm species (Fig. S1). Nevertheless, some variation could be detected. For instance, other palm species have two copies of therps19 gene located near the IRA/LSC and IRB/SSC junctions respectively, whereas coconut has only one copy of rps19 at the IRB/SSC junction. At the IRA/LSC junction we found a rps19-like sequence of 174 bp, which is likely a pseudogene judged from its shorter length compared to the regular rps19 gene (279 bp). We speculate that the pseudogenization of the rps19 at IRA/LSC junction is due to IR fluctuation in coconut cpDNA.
A comparative study among cpDNAs of six palm species (Table 5) indicated that coconut has the smallest cp genome (154,731 bp) and the shortest IRs (53,110 bp). The largest cp genome with the longest IRs is found in Phoenix (158,462 bp and 54,552 bp, respectively). Similarly to other cp genomes [2], [3], the palm cp genomes, including coconut, are all AT-rich. Graphical alignment showed that the IRs have both expanded and contracted during the evolution of the palm family, though dramatic changes were not detected (Fig. 2).
Figure 2. IR expansion into the LSC and SSC regions.
Comparison of IR boundaries among six palm species. Numbers in red denote distance between rpl22 and junction of LSC and IRB. Numbers in blue denote distance between rps19 and junction of LSC and IRA. Numbers in gray denote distance between psbA and junction of LSC and IRA.
Fluctuations of the IR regions have occurred sporadically during the evolutionary history of angiosperms [55]. Two of the most extreme cases are found in Pelargonium hortorum of the Geraniaceae and a group of legumes that includes pea and broad beans. The single IR region has expanded to 76 kb [46] in the former whereas one copy of the IRs is completely lost from cp genomes of the latter [1]. The structurally conserved feature of the IR regions is resistant to recombinational loss [56]. The presence of the IR regions may thus help to stabilize the cp genome. The most direct evidence for this suggestion is that more rearrangements occurred within the group of legumes that have lost a copy of IR than those that have not [57]. Another piece of evidence is the acceleration of synonymous substitution rates in the remaining copy of the duplicated region [56]. Consequently, we can infer that the evolutionary rates of cp genomes in the palm family are relatively mild, judging from the comparatively minor fluctuation of the IR regions.
Phylogenetic analysis and events of gene gain and loss
Our phylogenetic reconstruction built upon 47 protein-coding genes of cp sequences, rooted by Amborella, supported three major monophyletic groups: magnoliids, monocots, and eudicots (Fig. 3). Within monocots, Acorus (Acorales) diverged from other monocots first, followed by Colocasia (Alismatales), then by Cymbidium (Asparagales), which is sister to a clade that forms a monophyletic group of commelinids. The commelinids contain two sister clades. Within the first clade, Arecales group with the family Dasypogonaceae. In the second clade, Poales is sister to a subclade, which includes Zingiberales and Commelinales (Fig. 3). This topology is consistent with a phylogenetic study of commelinids based on 83 plastid genes [8]. Moreover, our inference of relationships within the Arecales is also congruent with a thorough study of the palm family using a supermatrix method with 16 data partition [58].
Figure 3. Phylogenetic tree of monocots.
Numbers above/below the branches are bootstrap value (only values higher than 50% are shown). Black square denotes rps19 duplication, gray square denotes rps19pseudogenization, white square denotes complete loss of duplicate rps19, and blue square denotes pseudo ycf1 and ndhF overlap.
We then mapped the related gene duplication and pseudogenization events onto the tree according to parsimony criteria. Our results indicate that the duplication of rps19 gene near the IRA/LSC junction likely occurred before the divergence of Asparagales from the remaining monocots, which consist of Arecales, a family (Dasypogonaceae) with indecisive order (Dasypogon and Kingia), Poales, Commelinales, and Zingiberales (Fig. 3). After the lineages differentiated, the duplicated rps19 eventually became a pseudogene independently in Cocosof the Arecales, Heliconia of the Zingiberales, and Nandina of the Ranunculales. It has been completely lost in Xiphidium of the Commelinales and Ceratophyllum of the Ceratophyllales (Fig. 3).
In monocots, the overlap between ndhF and pseudo ycf1 was found in a clade that contains Arecales and Dasypogonaceae. However, it was also found in Drimys of the Canellales and Chloranthus of the Chloranthales, both belong to the magnoliids. Following the parsimony rule, we concluded that the occurrence of the overlap between ndhF and pseudo ycf1 in monocots and magnoliids arose from three independent events.
In summary, we have presented here the first complete cp genome sequence from coconut palm. Although the cp genome of coconut is the smallest found so far among palms, it shares the same overall organization, gene content and repeat structure that have been observed with cpDNA sequenced from other palm species. Nevertheless, unique features were found for the coconut genome, including pseudogenization of rps19-like gene and an unusually high number of RNA editing sites. A closer relationship between coconut and oil palms than with date palm was supported by phylogenetic relationships among angiosperms. Our data will contribute to the growing number of molecular and genomic resources available for studying coconut palm biology.
Supporting Information
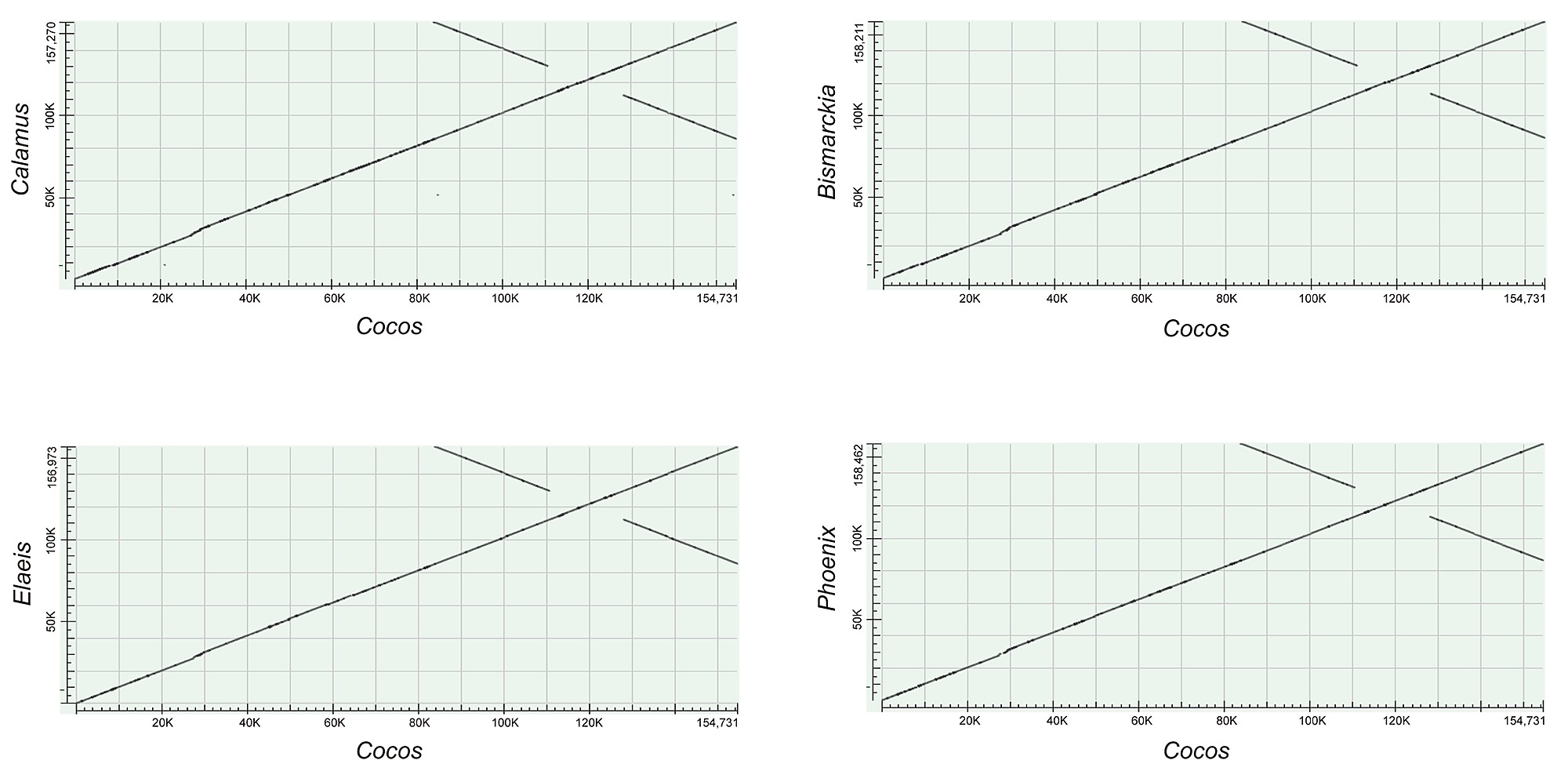
Figure S1.
Dot plot analysis. The cp genomes are nearly identical in the palm family.
doi:10.1371/journal.pone.0074736.s001
(TIF)
Acknowledgments
We thank Mr. Chi-Tai Lin, a local dwarf coconut breeder, from Hengchun peninsula in southern Taiwan for providing coconuts.
Author Contributions
Conceived and designed the experiments: YYH AJMM MM. Analyzed the data: YYH. Contributed reagents/materials/analysis tools: YYH. Wrote the paper: YYH MM.
References
- 1.Palmer J (1985) Comparative organization of chloroplast genomes. Annu Rev Genet 19: 325–354. doi: 10.1146/annurev.ge.19.120185.001545
- 2.Yang M, Zhang X, Liu G, Yin Y, Chen K, et al. (2010) The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PloS ONE 5: e12762. doi: 10.1371/journal.pone.0012762
- 3.Uthaipaisanwong P, Chanprasert J, Shearman JR, Sangsrakru D, Yoocha T, et al. (2012) Characterization of the chloroplast genome sequence of oil palm (Elaeis guineensis Jacq.). Gene 500: 172–180. doi: 10.1016/j.gene.2012.03.061
- 4.Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2011) Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol 28: 583–600. doi: 10.1093/molbev/msq229
- 5.Cai Z, Penaflor C, Kuehl J, Leebens-Mack J, Carlson J, et al. (2006) Complete plastid genome sequences of Drimys, Liriodendron, and Piper: implications for the phylogenetic relationships of magnoliids. BMC Evol Biol 6: 77.
- 6.Tangphatsornruang S, Sangsrakru D, Chanprasert J, Uthaipaisanwong P, Yoocha T, et al. (2010) The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput Pyrosequencing: structural organization and phylogenetic relationships. DNA Res 17: 11–22. doi: 10.1093/dnares/dsp025
- 7.Meerow A, Krueger R, Singh R, Low E-T, Ithnin M, et al.. (2012) Coconut, date, and oil palm genomics. In: Schnell RJ, Priyadarshan PM, Genomics of Tree Crops: Springer New York. 299–351.
- 8.Barrett CF, Davis JI, Leebens-Mack J, Conran JG, Stevenson DW (2013) Plastid genomes and deep relationships among the commelinid monocot angiosperms. Cladistics 29: 65–87. doi: 10.1111/j.1096-0031.2012.00418.x
- 9.Harries HC (1978) The evolution, dissemination and classification of Cocos nucifera L. Bot Rev 44: 265–319. doi: 10.1007/bf02957852
- 10.Gunn BF, Baudouin L, Olsen KM (2011) Independent origins of cultivated coconut (Cocos nucifera L.) in the Old World tropics. PLoS ONE 6: e21143. doi: 10.1371/journal.pone.0021143
- 11.Perera L, Russell JR, Provan J, Powell W (2003) Studying genetic relationships among coconut varieties/populations using microsatellite markers. Euphytica 132: 121–128. doi: 10.1023/a:1024696303261
- 12.Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci USA 81: 8014–8018. doi: 10.1073/pnas.81.24.8014
- 13.Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20: 3252–3255. doi: 10.1093/bioinformatics/bth352
- 14.Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl Acids Res 32: 1792. doi: 10.1093/nar/gkh340
- 15.Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucl Acids Res 33: W686–W689. doi: 10.1093/nar/gki366
- 16.Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucl Acids Res 27: 573–580. doi: 10.1093/nar/27.2.573
- 17.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, et al. (2000) Artemis: sequence visualization and annotation. Bioinformatics 16: 944–945. doi: 10.1093/bioinformatics/16.10.944
- 18.Mower JP (2009) The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucl Acids Res 37: W253–W259. doi: 10.1093/nar/gkp337
- 19.Qian J, Song J, Gao H, Zhu Y, Xu J, et al. (2013) The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 8: e57607. doi: 10.1371/journal.pone.0057607
- 20.Shimada H, Sugiura M (1991) Fine structural features of the chloroplast genome: comparison of the sequenced chloroplast genomes. Nucleic Acids Res 19: 983–995. doi: 10.1093/nar/19.5.983
- 21.Delannoy E, Fujii S, Colas des Francs-Small C, Brundrett M, Small I (2011) Rampant gene loss in the underground orchid Rhizanthella gardneri highlights evolutionary constraints on plastid genomes. Mol Biol Evol 28: 2077–2086. doi: 10.1093/molbev/msr028
- 22.Donaher N, Tanifuji G, Onodera NT, Malfatti SA, Chain PSG, et al. (2009) The complete plastid genome sequence of the secondarily non-photosynthetic alga Cryptomonas paramecium: reduction, compaction, and accelerated evolutionary rate. Genome Biol Evol. 1: 439–448. doi: 10.1093/gbe/evp047
- 23.Chen X, Kindle KL, Stern DB (1995) The initiation codon determines the efficiency but not the site of translation initiation in Chlamydomonas chloroplasts. Plant Cell 7: 1295–1305. doi: 10.2307/3870103
- 24.Rohde W, Gramstat A, Schmitz J, Tacke E, Prüfer D (1994) Plant viruses as model systems for the study of non-canonical translation mechanisms in higher plants. J Gen Virol 75: 2141–2149. doi: 10.1099/0022-1317-75-9-2141
- 25.Goremykin VV, Hirsch-Ernst KI, Wölfl S, Hellwig FH (2003) Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol Biol Evol 20: 1499–1505. doi: 10.1093/molbev/msg159
- 26.Raubeson L, Peery R, Chumley T, Dziubek C, Fourcade H, et al. (2007) Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8: 1–27. doi: 10.1186/1471-2164-8-174
- 27.Moore MJ, Bell CD, Soltis PS, Soltis DE (2007) Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci USA 104: 19363–19368. doi: 10.1073/pnas.0708072104
- 28.Ahmed I, Biggs PJ, Matthews PJ, Collins LJ, Hendy MD, et al. (2012) Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol 4: 1316–1323. doi: 10.1093/gbe/evs110
- 29.Yang JB, Tang M, Li HT, Zhang ZR, Li DZ (2013) Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol Biol 13: 84. doi: 10.1186/1471-2148-13-84
- 30.Jansen RK, Cai Z, Raubeson LA, Daniell H, dePamphilis CW, et al. (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA 104: 19369–19374. doi: 10.1073/pnas.0709121104
- 31.Hansen DR, Dastidar SG, Cai Z, Penaflor C, Kuehl JV, et al. (2007) Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea(Dioscoreaceae), and Illicium (Schisandraceae). Mol Phylogenet Evol 45: 547–563. doi: 10.1016/j.ympev.2007.06.004
- 32.Moore MJ, Dhingra A, Soltis PS, Shaw R, Farmerie WG, et al. (2006) Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol 6: 17. doi: 10.1186/1471-2229-6-17
- 33.Zandueta-Criado A, Bock R (2004) Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res 32: 542–550. doi: 10.1093/nar/gkh217
- 34.Sasaki T, Yukawa Y, Miyamoto T, Obokata J, Sugiura M (2003) Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol 20: 1028–1035. doi: 10.1093/molbev/msg098
- 35.Guisinger MM, Chumley TW, Kuehl JV, Boore JL, Jansen RK (2010) Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J Mol Evol 70: 149–166. doi: 10.1007/s00239-009-9317-3
- 36.Gray BN, Ahner BA, Hanson MR (2009) Extensive homologous recombination between introduced and native regulatory plastid DNA elements in transplastomic plants. Transgenic Res 18: 559–572. doi: 10.1007/s11248-009-9246-3
- 37.Rogalski M, Ruf S, Bock R (2006) Tobacco plastid ribosomal protein S18 is essential for cell survival. Nucl Acids Res 34: 4537–4545. doi: 10.1093/nar/gkl634
- 38.Marechal A, Parent JS, Veronneau-Lafortune F, Joyeux A, Lang BF, et al. (2009) Whirly proteins maintain plastid genome stability in Arabidopsis. Proc Natl Acad Sci U S A 106: 14693–14698. doi: 10.1073/pnas.0901710106
- 39.Zhang Y-J, Ma P-F, Li D-Z (2011) High-throughput sequencing of six bamboo chloroplast genomes: phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 6: e20596. doi: 10.1371/journal.pone.0020596
- 40.Saski C, Lee SB, Fjellheim S, Guda C, Jansen RK, et al. (2007) Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes. Theor Appl Genet 115: 571–590. doi: 10.1007/s00122-007-0567-4
- 41.Bausher M, Singh N, Lee S-B, Jansen R, Daniell H (2006) The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol 6: 21.
- 42.Daniell H, Lee S-B, Grevich J, Saski C, Quesada-Vargas T, et al. (2006) Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor Appl Genet 112: 1503–1518. doi: 10.1007/s00122-006-0254-x
- 43.Lee S-B, Kaittanis C, Jansen R, Hostetler J, Tallon L, et al. (2006) The complete chloroplast genome sequence of Gossypium hirsutum: organization and phylogenetic relationships to other angiosperms. BMC Genomics 7: 1–12.
- 44.Cai Z, Guisinger M, Kim H, Ruck E, Blazier J, et al.. (2008) Extensive reorganization of the plastid genome of Trifolium subterraneum (Fabaceae) is associated with numerous repeated sequences and novel DNA insertions. J Mol Evol.
- 45.Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, et al. (2006) The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol 23: 2175–2190. doi: 10.1093/molbev/msl089
- 46.Palmer J, Nugent J, Herbon L (1987) Unusual structure of geranium chloroplast DNA: A triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc Natl Acad Sci USA 84: 769–773. doi: 10.1073/pnas.84.3.769
- 47.Chen H, Deng L, Jiang Y, Lu P, Yu J (2011) RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J Integr Plant Biol 53: 961–970. doi: 10.1111/j.1744-7909.2011.01082.x
- 48.Wakasugi T, Hirose T, Horihata M, Tsudzuki T, Kössel H, et al. (1996) Creation of a novel protein-coding region at the RNA level in black pine chloroplasts: the pattern of RNA editing in the gymnosperm chloroplast is different from that in angiosperms. Proc Natl Acad Sci USA 93: 8766–8770. doi: 10.1073/pnas.93.16.8766
- 49.Gray M, Covello P (1993) RNA editing in plant mitochondria and chloroplasts. The FASEB Journal 7: 64–71. doi: 10.1038/341662a0
- 50.Tillich M, Lehwark P, Morton BR, Maier UG (2006) The evolution of chloroplast RNA editing. Mol Biol Evol 23: 1912–1921. doi: 10.1093/molbev/msl054
- 51.Corneille S, Lutz K, Maliga P (2000) Conservation of RNA editing between rice and maize plastids: are most editing events dispensable? Mol Gen Genet 264: 419–424. doi: 10.1007/s004380000295
- 52.Lutz KA, Maliga P (2001) Lack of conservation of editing sites in mRNAs that encode subunits of the NAD(P)H dehydrogenase complex in plastids and mitochondria of Arabidopsis thaliana. Curr Genet 40: 214–219. doi: 10.1007/s002940100242
- 53.Hirose T, Kusumegi T, Tsudzuki T, Sugiura M (1999) RNA editing sites in tobacco chloroplast transcripts: editing as a possible regulator of chloroplast RNA polymerase activity. Mol Gen Genet 262: 462–467. doi: 10.1007/s004380051106
- 54.Guzowska-Nowowiejska M, Fiedorowicz E, Pląder W (2009) Cucumber, melon, pumpkin, and squash: Are rules of editing in flowering plants chloroplast genes so well known indeed? Gene 434: 1–8. doi: 10.1016/j.gene.2008.12.017
- 55.Goulding S, Wolfe K, Olmstead R, Morden C (1996) Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet 252: 195–206. doi: 10.1007/bf02173220
- 56.Perry AS, Wolfe KH (2002) Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J Mol Evol 55: 501–508. doi: 10.1007/s00239-002-2333-y
- 57.Palmer JD, Thompson WF (1982) Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 29: 537–550. doi: 10.1016/0092-8674(82)90170-2
- 58.Baker WJ, Savolainen V, Asmussen-Lange CB, Chase MW, Dransfield J, et al. (2009) Complete generic-level phylogenetic analyses of palms (Arecaceae) with comparisons of supertree and supermatrix approaches. Syst Biol 58: 240–256.
- 59.Martin G, Baurens F-C, Cardi C, Aury J-M, D'Hont A (2013) The complete chloroplast genome of banana (Musa acuminata, Zingiberales): insight into plastid monocotyledon evolution. PLoS ONE 8: e67350. doi: 10.1371/journal.pone.0067350
For further details log on website :
http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0122123









No comments:
Post a Comment